


Primary bile acid synthesis disorders
What are BASD?
Bile acid synthesis disorders (BASD) are rare genetic conditions that can present as cholestasis, neurologic disease, or fat-soluble-vitamin deficiencies. They are responsible for 1-2% of cases of neonatal cholestasis. (1)
Primary BASD are rare causes of liver disease in children that can occur at any age (from birth to adolescence). However, the possibility of diagnosing these diseases in adults should not be ruled out. They usually present as cholestatic jaundice and/or liver failure. (3) Early diagnosis of these disorders is essential.
The two most frequent ones responsible for chronic liver disease are: (2)
3ß-hydroxy-Δ5-C27-steroid oxidoreductase (or dehydrogenase / isomerase) deficiency; may also be called 3ß-HSD deficiency, or BAS defect type 1.
Δ4-3-oxosteroid-5ß reductase deficiency; may also be called Δ4-3-oxoR, or 5ß-reductase deficiency, or BAS defect type 2.
They are caused by the lack of one of the two enzymes, 3ß-HSD or Δ4-3-oxoR, which are involved in the transformation of cholesterol into primary bile acids: cholic acid (CA) and chenodeoxycholic acid (CDCA). When they are missing, cholesterol transformation is incomplete and leads to the absence of primary bile acids and accumulation of toxic intermediates in the liver causing cholestasis, then liver cirrhosis or progressive and irreversible liver failure. (2)
In Europe, the prevalence of 3ß-HSD and Δ4-3-oxoR deficiencies is at least 1.13 cases per 10 million people: (3)
0.99 per 10 million people for 3ß-HSD deficiency,
0.14 per 10 million people for Δ4-3-oxoR deficiency.
However, it is possible that these prevalences may be underestimated. (3)
Symptoms and Diagnosis
These diseases are autosomal recessive; for them to develop, a person has to inherit two mutated genes (a mutated gene from the mother and a mutated gene from the father).
Parent consanguinity increases the risk of carrying the disease and developing it.
These diseases may be suspected based on a combination of clinical signs, laboratory results, the patient's family history, and liver histology findings. Observed together, they should lead to a specific urine test and then a genetic test to confirm the disease.
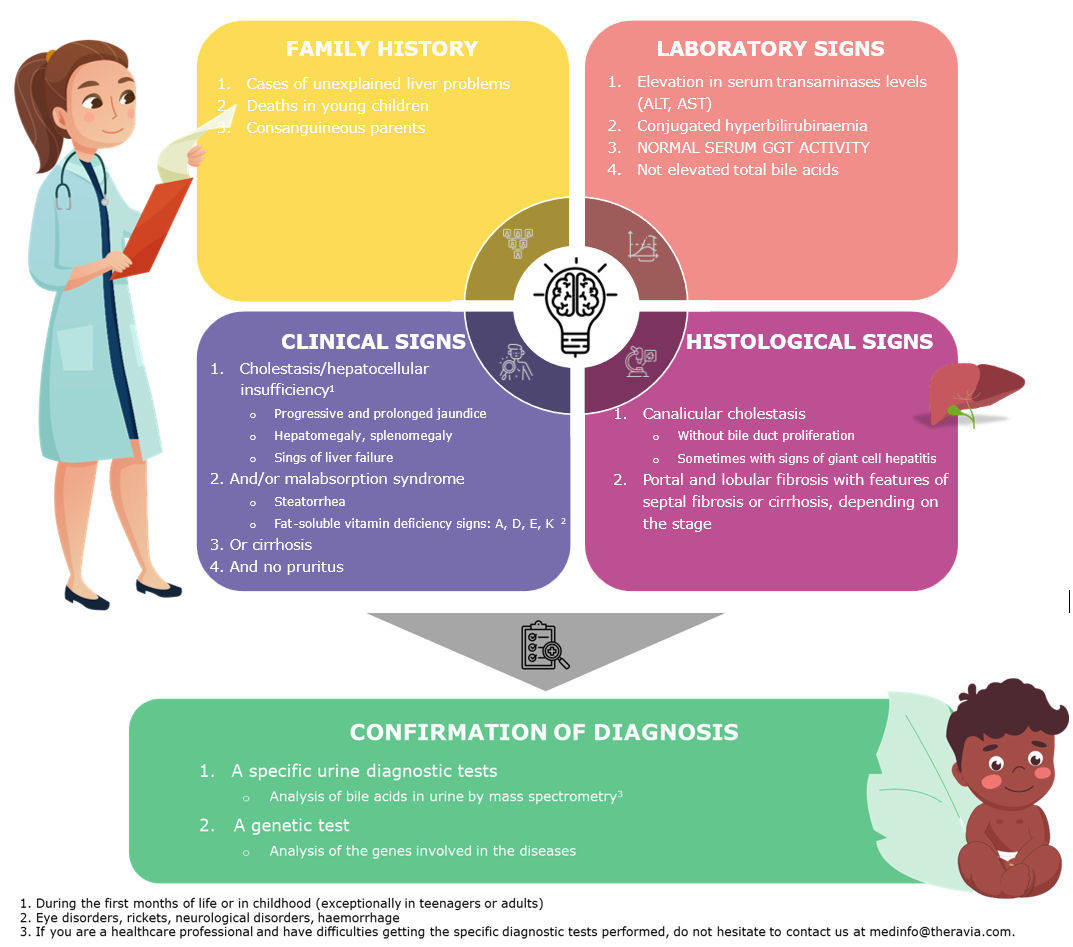
Diagnosis can be difficult because there are no specific clinical features or biomarkers allowing the specific identification of BASDs. However, most BASD patients present as cholestasis with (1;4)
Normal or low total serum bile acid concentrations
Normal γ-glutamyl transpeptidase concentrations
No pruritus
Patients with Δ4-3-oxoR deficiency are similar to patients with 3ß-HSD deficiency. However, the average diagnosis is 3 months in patients with Δ4-3-oxoR versus 3 months to 14 years in patients with 3ß-HSD deficiency. (1-2)
They also tend to have more severe liver disease than patients with 3ß-HSD deficiency and more rapid progression to cirrhosis and death without intervention. (5)
Management of the disease
Early diagnosis is important because these disorders can be treated. Without treatment, there is a 50% mortality rate of Δ4-3-oxoR deficiency in infants for whom diagnosis is delayed. (1-6)
Treatment is based on supplementation with primary bile acids. Treatment should be initiated in a specialised environment, where the patient should also be monitored.(2) Follow-up includes regular visits and regular blood and urine tests.
Living with the disease
It is important to take medical treatment continuously because stopping it may lead to the reappearance of symptoms and further deterioration of the liver.
Resources:
Association Maladie Foie Enfant – Child Liver Disease Association - https://amfe.fr/
Filfoie – French network for rare liver diseases - https://www.filfoie.com/
CLDF – Chronic Liver Disease Foundation - https://www.chronicliverdisease.org/
References:
Sundaram SS, Bove KE, Lovell MA, Sokol RJ. Mechanisms of disease: Inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol 2008;5:456-68.
Protocole national de diagnostic et de soins : Déficits de synthèse des acides biliaires primaires. Centre de Référence Coordonnateur de l’Atrésie des Voies Biliaires et des Cholestases Génétiques; 2019
J Jahnel, E Zohrer, B Fischler, L D’Antiga, D Debray, A Dezsofi, et al. Attempt to determine the prevalence of two inborn errors of primary bile acid synthesis: results of a european survey. JPGN, 2017 (64), 864–868.
Bile acid synthesis disorders. NORD: National Organization for Rare Disorders, 2017. (Accessed April, 2020, at https://rarediseases org/rare-diseases/bile-acid-synthesis-disorders/.)
Heubi JE, Setchell KDR, Bove KE. Inborn errors of bile acid metabolism. Clin Liver Dis 2018;22:671-87
K E Bove, J E Heubi, W F Balistreri , K D R Setchell. Bile acid synthetisis defects and liver disease : a comprehensive review. Pediatric and Developmental Pathology; 2004 (7), 315-334.